输入与输出
本文将介绍ChloroScan的输入和输出,并详细介绍工作流程中的每个步骤。我们还将为用户提供一些检查数据和结果的提示。
- 总体而言,ChloroScan 接受以下输入:
以 fasta 格式的宏基因组组装(必需):通过
--Inputs-assembly参数配置。测序深度表格(必需
tsv,与下一个选项互斥):通过--Inputs-depth-profile参数配置。存储排序后的 bam 文件的目录(必需):通过
--Inputs-alignment参数配置。批次名称(必需,仅用于在并行运行时区分不同作业以避免覆盖):通过
--Inputs-batch-name参数配置。
通常对于那些第一次进行分析的数据( de novo assembly ),我们建议使用 bam 目录,但如果您想使用现有的深度谱重新运行分析(可能调整参数),也可以使用它。
ChloroScan 的输出目录结构如下:
chloroscan_output
├── logging_info
└── working
├── binny
│ ├── bins
│ ├── intermediary
│ │ └── mantis_out
│ ├── job.errs.outs
│ ├── logs
│ └── tmp
├── call_contig_depth
├── CAT
├── cds-extraction
│ ├── cds
│ └── faa
├── corgi
├── FragGeneScanRs
│ └── GFFs
├── refined_bins
├── summary
└── visualizations
1. 输入数据
- 宏基因组组装 (.fasta)
ChloroScan 接受宏基因组组装的contigs,其中包含样品中各种基因组片段,包括细菌、古菌、真核生物和病毒。 我们目前测试的组装类型仅限于短读长,您可以使用 megahit 或 metaSPAdes 生成。我们计划在未来测试长读长的结果。 在分析时,请确保您的 contigs 具有不重复的 id,并且这些 id 与 bam 文件中的 id 一致。例如,如果您的 fasta 文件中的 contig id 是 “k141_1000298”,则 bam 文件中的对应 contig id 也应为 “k141_1000298”。 通常,平均 contig 较长的组装会产生更好的结果,但这也取决于组装的质量。
- BAM文件
通过将测序的reads比对回组装的contigs,您可以获得每个contig在样品中的测序深度。对于短读长,minimap2(短读模式)、bowtie2 和 bwa-mem 效果都不错。
2. Corgi预测
Corgi 的第一个步骤:contig 分类仅需要您的 input-contigs.fasta 文件。
程序首先将每个 contig 分类为五个基于 RefSeq 的类别:叶绿体、线粒体、原核生物、真核生物和未知。
然后输出结果:分类结果 corgi-prediction.csv,以及分离的 plastids.fasta 文件将生成在 output/working/corgi 目录中。
分类结果 corgi-prediction.csv 是一个表格文本文件,包含每个 contig 的分类结果、每个类别的后验概率以及 contig 的长度。它看起来如下所示:
file,accession,prediction,probability,original_id,description,Nuclear,Mitochondrion,Plastid,Plasmid,Nuclear/Bacteria,Nuclear/Archaea,Nuclear/Eukaryota,Nuclear/Viruses,Mitochondrion/Eukaryota,Plastid/Eukaryota,Plasmid/Bacteria,Plasmid/Archaea,Plasmid/Eukaryota
assembly.formatted.fa,k97_110,Nuclear/Bacteria,0.5487518906593323,k97_110,k97_110,0.5688353,0.00050010456,0.016087921,0.41457665,0.5487519,0.0055856705,0.006795563,0.007702232,0.0,0.0,0.41455323,2.3394885e-05,4.0987683e-08
assembly.formatted.fa,k97_2859999,Nuclear/Bacteria,0.8199571371078491,k97_2859999,k97_2859999,0.8871814,0.001272265,0.07066683,0.040879533,0.81995714,0.0014582742,0.00072807295,0.06503786,0.0,0.0,0.04087944,9.103693e-08,1.9135307e-09
assembly.formatted.fa,k97_2451439,Plastid,0.8210093379020691,k97_2451439,k97_2451439,0.11955466,0.037155744,0.82100934,0.022280276,0.04770646,0.0004202699,0.04415435,0.027273588,0.0,0.0,0.022280134,1.3834504e-07,2.588508e-09
要配置此步骤:
--corgi-pthreshold: 将 contig 分类为五个类别之一的后验概率阈值。默认值为 0.5。
--corgi-min-length: 要考虑的 contig 的最小长度。默认值为 1000bp。
--corgi-save-filter: 运行 corgi 的默认过滤步骤以分离叶绿体 contig。我们不推荐您这么做,因为运行时间较长。
--corgi-batch-size: 推理contig的物种分类时的批次大小,默认值为 1。
3. binny分箱结果
此步骤的输入是来自 Corgi 的 plastids.fasta 文件和以表格形式表示的测序平均深度,第一列是 contig id,其余列是每个样品的平均深度。
注意: 如果用户未指定测序深度表格,ChloroScan 会直接使用 binny 的代码生成深度谱。其路径为output/working/depth_profile.txt。
输出目录为 output/working/binny。目录 bins 包含最终的叶绿体MAGs,格式为 fasta。
由于 binny 是一个 snakemake 工作流,其他中间结果也会保留在 ChloroScan 中。
如下展示的是一个binny运行的中间结果目录结构:
intermediary ├── annotation_CDS_RNA_hmms_checkm.gff ├── annotation.filt.gff ├── assembly.contig_depth.txt ├── assembly.formatted.fa ├── iteration_1_contigs2clusters.tsv ├── iteration_2_contigs2clusters.tsv ├── iteration_3_contigs2clusters.tsv ├── mantis_out │ ├── consensus_annotation.tsv │ ├── integrated_annotation.tsv │ ├── Mantis.out │ └── output_annotation.tsv ├── prokka.faa ├── prokka.ffn ├── prokka.fna ├── prokka.fsa ├── prokka.gff ├── prokka.log ├── prokka.tbl ├── prokka.tsv └── prokka.txt
- 简单来说,您需要了解以下内容:
输入的 contigs 和以表格形式表示的测序深度分别在
assembly.formatted.fa和assembly.contig_depth.txt中。从 contigs 预测的开放阅读框存储在文件
annotation_CDS_RNA_hmms_checkm.gff中。
- 要配置此步骤:
--binning-clustering-hdbscan-min-sample-range: 运行 HDBSCAN 聚类时最小样本参数的范围,默认值为 1,5,10.--binning-bin-quality-purity: 保留 bin 的最小纯度阈值,默认值为 90(最大可接受污染程度为10%)。--binning-bin-quality-start-completeness: 保留 bin 的完整性起始值,默认值为 92.5(最小可接受完整度为92.5%)。--binning-bin-quality-min-completeness: 保留 bin 的最小完整性阈值,默认值为 50(最小可接受完整度为50%)。--binning-outputdir: 存储 binny 结果的输出目录,然后再移动到 ChloroScan 输出,默认值为output/working/binny。--binning-universal-length-cutoff: 应用于过滤 contigs 以进行标记基因预测和聚类的长度截止值,默认值为 1500bp。--binning-clustering-epsilon-range: HDBSCAN 的 epsilon 取值范围。我们不建议更改此值,因为效果不大,默认值为 0.250,0.000。
有关 HDBSCAN 的更多信息,请参见 sklearn 文档: https://scikit-learn.org/stable/modules/generated/sklearn.cluster.HDBSCAN.html.
4. CAT
输入是中间目录中的 assembly.formatted.fasta 文件。它存储了长度超过 500bp 的推定叶绿体 contigs。
总体上,唯一有用的信息在 out.CAT.contig2classification.txt 中,它是一个表格文本文件,存储了每个 contig 的分类结果、推定谱系以及每个分类单元的相应分数。
这个表格看起来如下所示:
# contig classification reason lineage lineage scores
k141_1000298 no taxid assigned no hits to database
k141_1000835 taxid assigned based on 2/2 ORFs 1;131567;2;1224;28211;54526 1.00;1.00;1.00;1.00;1.00;1.00
k141_1001040 taxid assigned based on 2/2 ORFs 1;131567;2;1224;28211;766;1699067;2026788 1.00;1.00;1.00;1.00;1.00;1.00;1.00;1.00
k141_1002229 taxid assigned based on 2/3 ORFs 1;131567;2 1.00;1.00;1.00
...
- 要配置此步骤:
--cat-database: CAT 数据库的路径(db目录),我们参考最新 CAT 数据库发布的目录结构。--cat-taxonomy: 分类文件的路径(taxonomy/tax目录)。
5. 总结模块
此步骤用于总结包括 binning 和 CAT 结果在内的元数据。它从之前的作业中获取输入以进行总结:
contigs 的基本信息,如 GC 含量、长度、每个样本的平均深度。
binning 结果:每个 contig 的 bin id。未分配的 contigs 显示为 NaN。
CAT 结果:每个 contig 的分类谱系。
原始输入的contigs序列。
本模块的输出表格就像如下的样子:
contig id GC contents contig depth contig length Taxon per Contig markers on the contig Contig2Bin contig sequence
contig_1 0.35 10.5 5000 1;131567;2;1224;28211;54526 atpA,atpB bin1 ATGCGT...
contig_2 0.40 20.0 3500 1;131567;2;1224;28211;766;1699067;2026788 psaA,psaC,rbcL bin2 ATGCGT...
contig_3 0.38 15.0 4000 1;131567;2; rpoC2,rpl4,rpl6 bin3 ATGCGT...
输出的 summary_table.tsv 是一个表格文本文件,存储了每个 contig 的所有上述信息。
5. “润色”模块
虽然在 binning 步骤中,binny 已经在保持 bins 纯度方面做了大量工作,但我们仍然发现它容易受到片段化 contigs 的影响,将污染附加到 bins 中。因此,我们在总结模块之后添加了一个“润色”步骤,以帮助用户检查这些 contigs 是否具有可疑的分类身份,并将它们从 bins 中移除。
为了向用户发出通知,我们在此报告了 binny 未预测标记基因且 CAT 分类不明确的 contigs(即非真核生物的或未被分类的), 例如“unclassified”、 “cellular organisms”、“bacteria”等。
另一种帮助用户的方法是运行 anvi’o interactive 模式,以查看 contigs 是否在系统发育图中形成均匀的簇。进一步指南:https://astrobiomike.github.io/genomics/metagen_anvio.
这个步骤的输入是上一步的 summary_table.tsv。输出目录为 output/working/refined_bins。我们还将信息总结到 output/working/refinement_contig_summary.txt 中,以报告哪个 bin 中的哪个 contig 具有可疑身份。
6. 预测开放阅读框和基因蛋白序列
对于每个 bin,我们通过 FragGeneScanRs 预测其编码的核苷酸序列和蛋白质序列。
输出位于 output/working/cds-extraction。目录 cds 包含 fna 格式的核苷酸序列,而 faa 包含 faa 格式的蛋白质序列。
- 我们使用以下参数配置 FragGeneScanRs:
-t illumina_5: 使用 Illumina 错误模型,错误率为 0.5%,适用于宏基因组数据。
7. 可视化
此步骤生成一些可视化图表,供用户快速浏览其数据。
对于样本中的所有 MAG,我们绘制它们的 GC 含量与对数转换后的汇总平均深度的关系。汇总平均深度是指对每个 contig 在每个样本中的深度进行求和。输出位于 output/working/visualizations/Scatter_GCLogDepth.png。
- 示例:
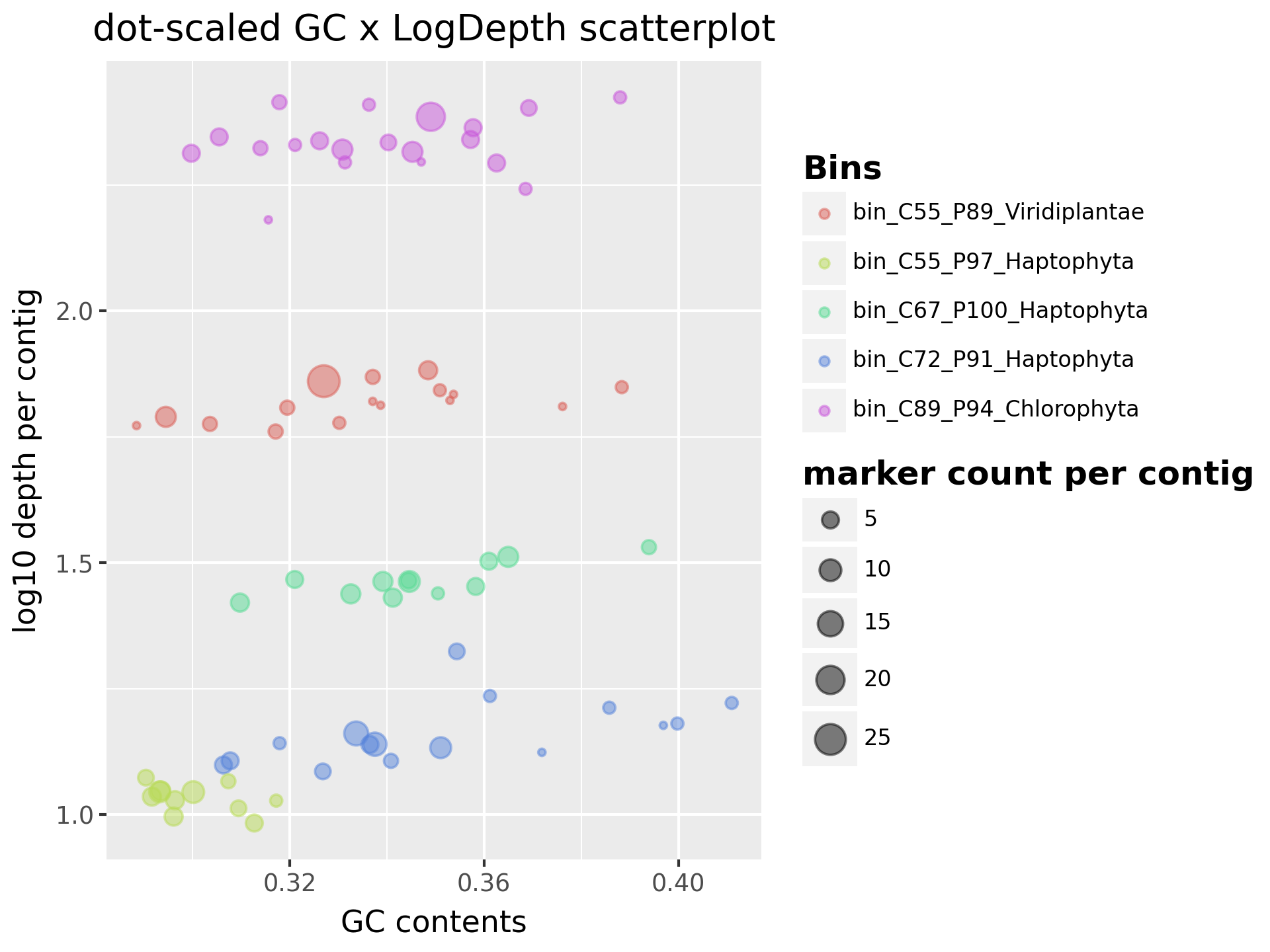
为了有效区分深度,我们对汇总平均深度进行了对数转换。正如示例所示,预期的良好 bins 应该具有均匀的深度和较少变化的 GC 含量,因此在图中形成一个紧密的簇。另一方面,具有更变量深度和 GC 含量的 bins 可能表明存在污染或错误分箱,因此在图中形成一个更分散的簇。
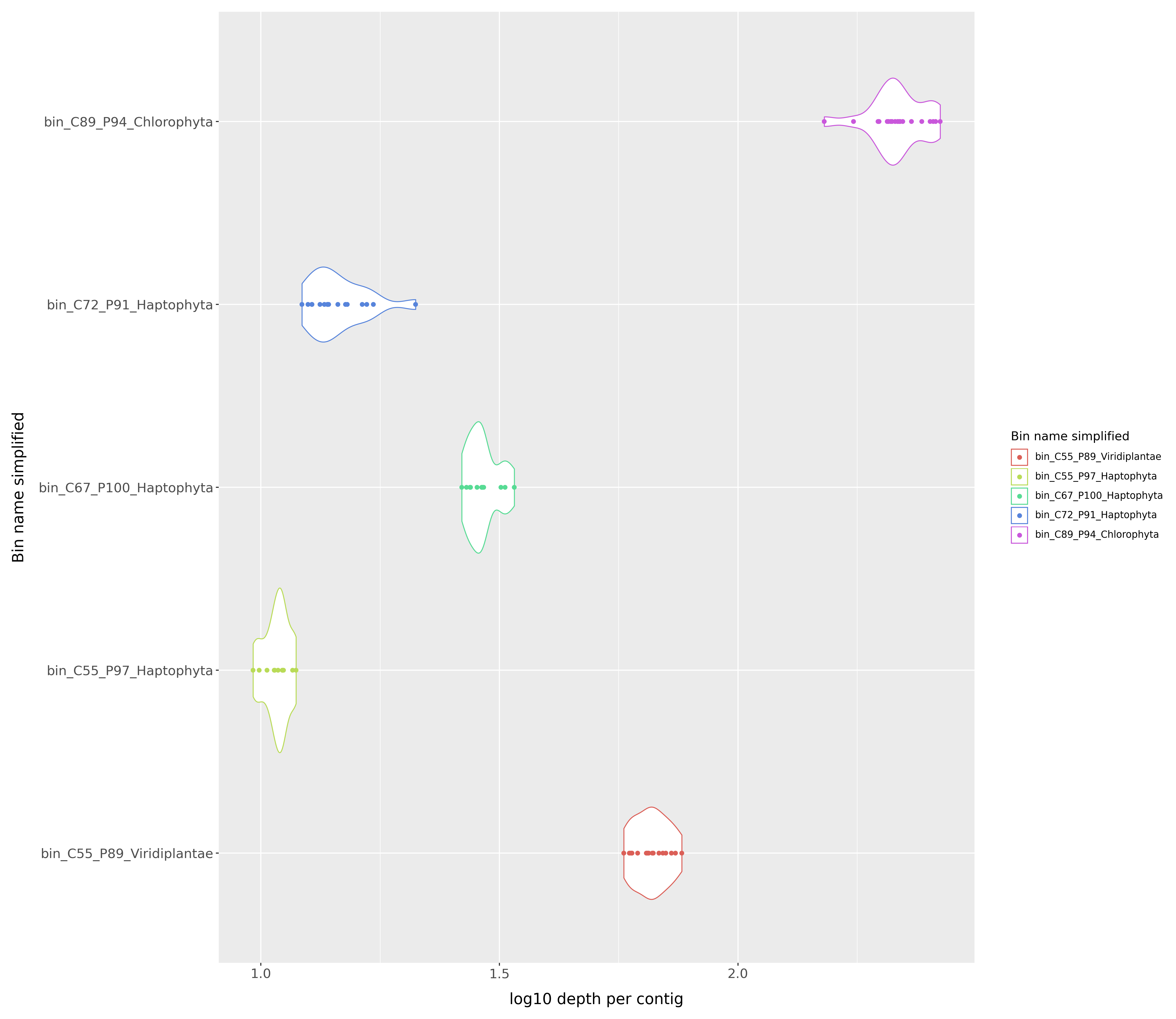
我们还生成了一个小提琴图,显示样本中所有 MAG 的汇总平均深度的分布。输出位于 output/working/visualizations。文件 depth_distribution.png 是一个小提琴图,显示样本中所有 MAG 的汇总平均深度的分布。
- 示例:
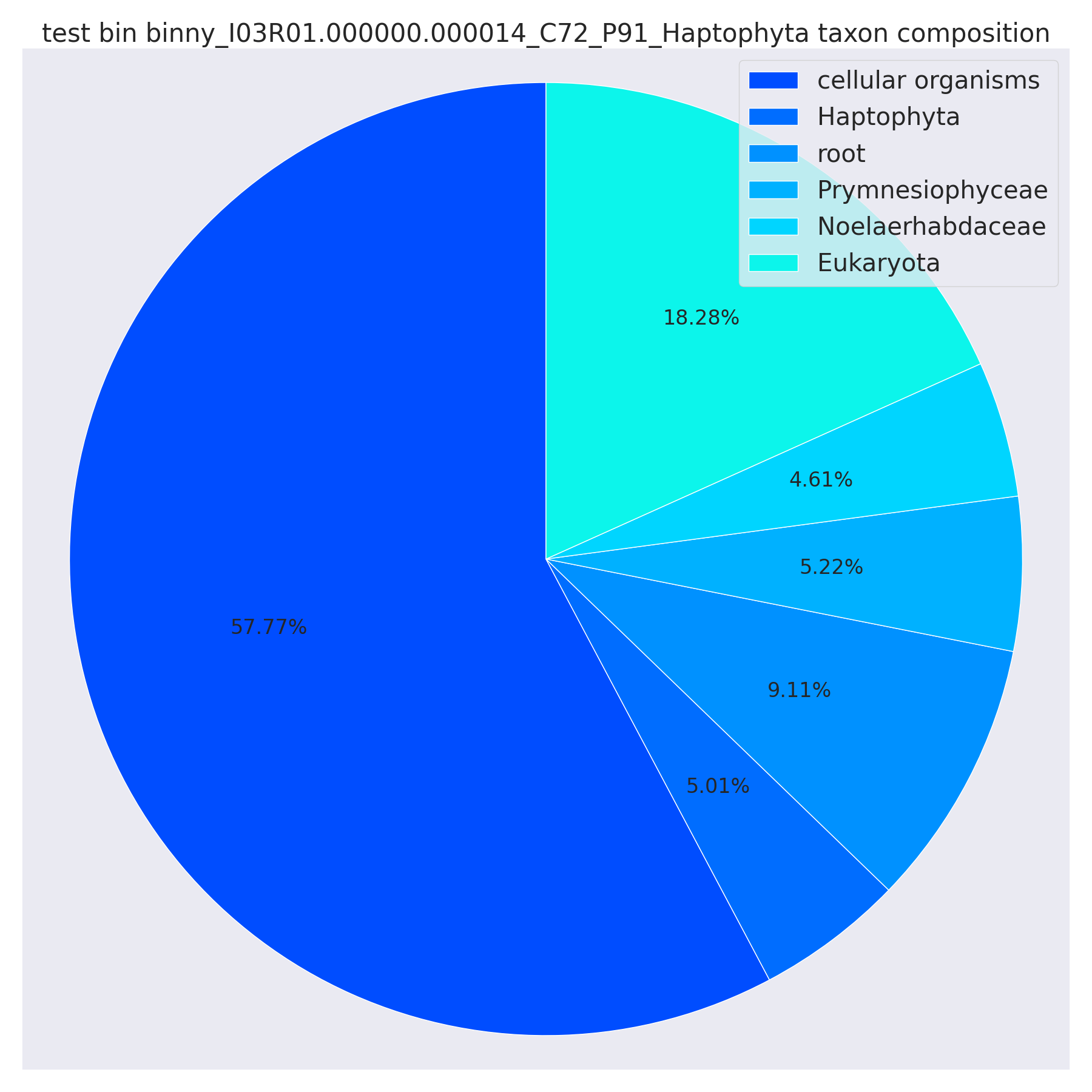
对于每个 bin,我们绘制其 contig 级别的分类组成饼图。文件命名为 {Batch_name}_{bin_id}_taxonomy_composition.png。 注意,CAT 可能在一个饼图中有多个分类单元,这些单元大致指向 bin 的一个分类(在这个例子中是 Haptophyte),其中一些 contigs 在更细的水平上被识别,而一些 contigs 在更粗的水平上被识别。未来参考基因组的深度采样的改进可能会有所帮助。
- 示例:
我们还为输入的宏基因组准备了一个 krona 图,用于可视化群落级别的分类分布,输出文件为指定输出目录下的 Krona.html。它将显示每个推定叶绿体 contig 的 CAT 预测分类。
- 示例:

8. 总结
- ChloroScan 的最重要输出如下:
output/corgi/plastid.fasta,存储预测的叶绿体 contigs 的文件。output/binny/bins,存储 binny 聚类的叶绿体 MAGs 的目录,格式为 fasta。output/summary/cross_ref.tsv,存储每个 contig 的元数据的表格文本文件,包括 binning 和 CAT 结果,以及原始序列的覆盖度和 GC 含量。output/visualizations,存储用于快速查看 MAGs 的图表的目录。output/cds-extraction,存储预测的 ORFs 和氨基酸序列的目录,可用于系统发育分析。